


 武大主页
武大主页 武大邮箱
武大邮箱 信息门户
信息门户 English
English
转自武大新闻网:新闻网讯(通讯员高妍)近日,高等研究院教授孔望清课题组在镍催化构建手性4-位偕二氟烯基取代吡咯烷酮方面研究取得新进展,研究成果发表在国际期刊Angew.Chem.Int.Ed.(《德国应用化学》)上。
论文题为“Nickel-Catalyzed Defluorinative Asymmetric Cyclization of Fluoroalkyl-Substituted 1,6-Enynes for the Synthesis of Seletracetam”(《镍催化1,6-烯炔环化脱氟反应构建手性4-位偕二氟烯基取代吡咯烷酮》)。高等研究院博士后王快,博士陈嘉昌、刘文峰为论文共同第一作者,孔望清为论文通讯作者。
吡咯烷酮骨架结构广泛存在于具有生物活性的天然产物中,同时也是合成具有药用价值化合物的重要前体。4-位偕二氟烯基取代吡咯烷酮化合物因其独特的结构,被广泛应用于医药开发中,例如Seletracetam在继发性和遗传性癫痫发作模型中具有强效的抑制癫痫发作的活性(图1)。目前其不对称合成主要依赖于手性拆分,因此发展合成手性4-位偕二氟烯基取代吡咯烷酮,特别是治疗癫痫药物Seletracetam的不对称合成方法学具有非常重要的意义。
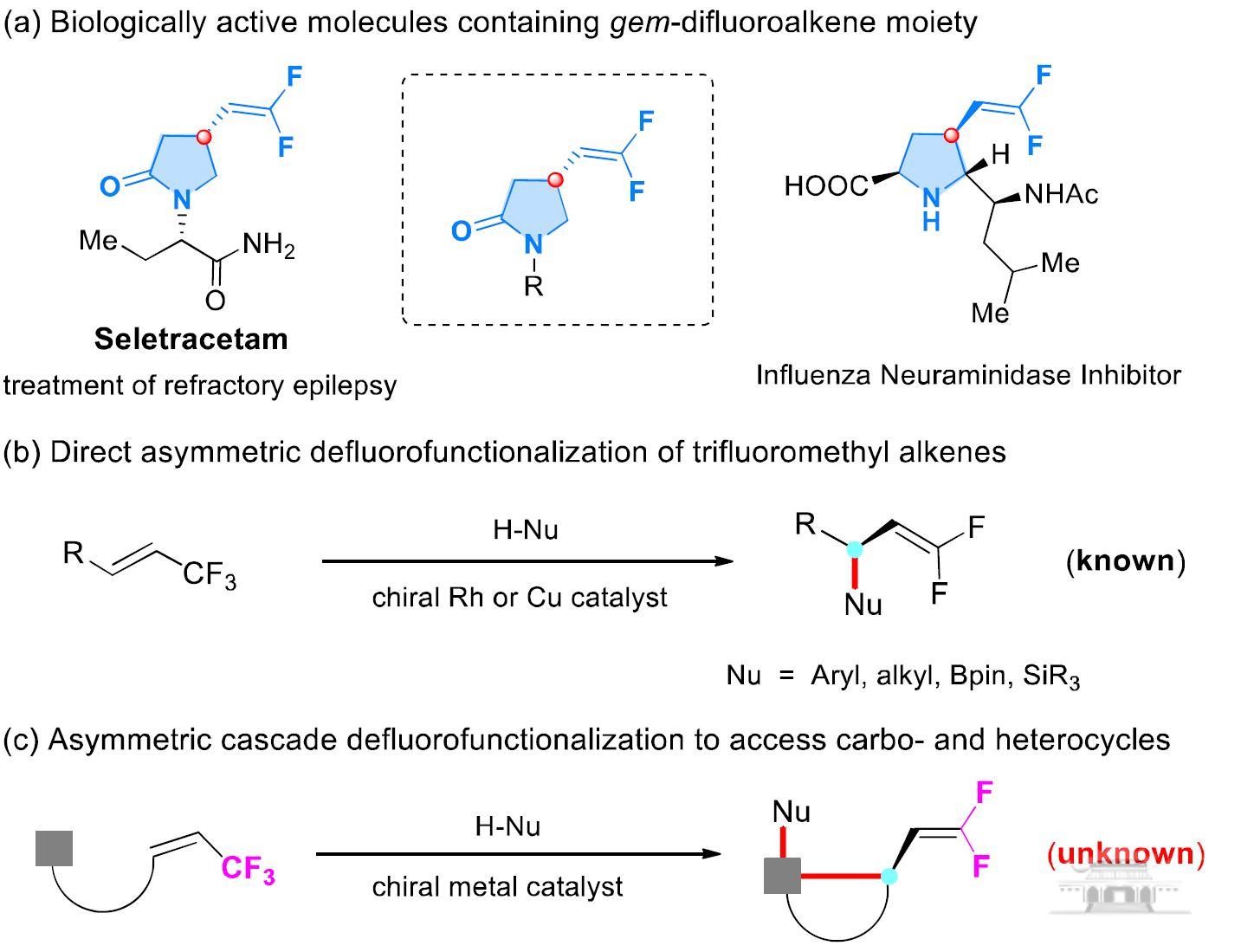
图1 含有4-位偕二氟烯基取代吡咯烷酮骨架结构的药物活性分子
孔望清课题组长期致力于镍催化不对称环化构建碳杂环的研究。为了解决Seletracetam的不对称合成问题,他们设计了通过三氟甲基取代的1,6-烯炔的加成/环化脱氟反应,来立体选择性构建4-位偕二氟烯基取代吡咯烷酮化合物(图2)。但实现这一反应仍有诸多挑战:一是化学选择性(碳碳三键和碳碳双键的反应选择性);二为区域选择性(碳碳三键反应时的位点选择性);三是Z/E选择性的调控(烯基镍物种经常会发生顺反异构化);四是立体选择性的控制。
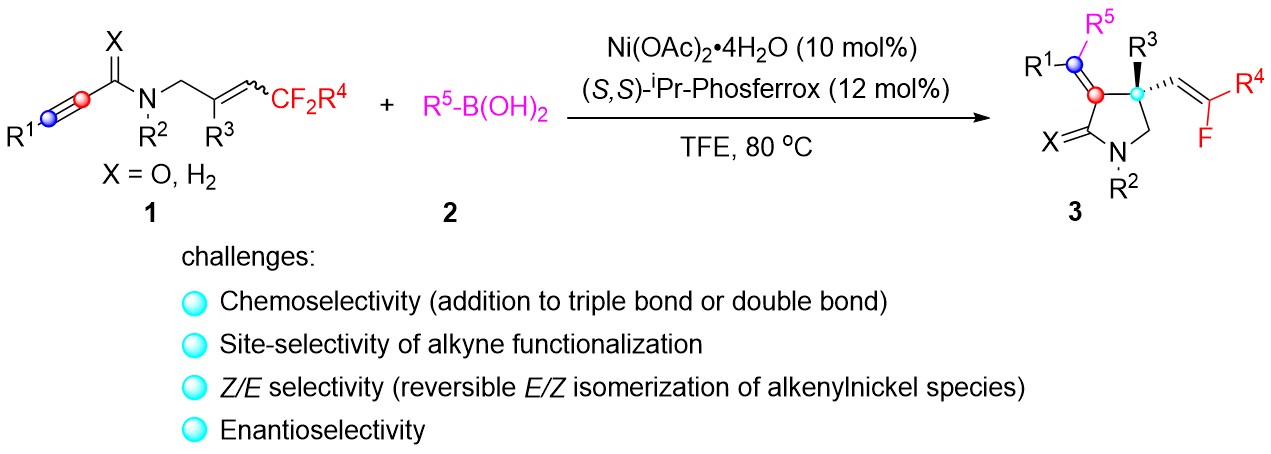
图2 镍催化1.6-烯炔不对称环化脱氟反应
经过大量实验课题组开发了一类镍催化手性4-位偕二氟烯基取代吡咯烷的合成方法,该方法可适用于多种含不同取代基的三氟甲基烯烃取代的1,6-烯炔与苯硼酸,原料廉价易得,是一种高效、绿色、廉价合成手性4-位偕二氟烯基取代吡咯烷酮的方法。并通过转化实现了手性seletracetam分子关键中间体(R)-4-(二氟乙烯基)-吡咯烷-2-酮的合成,体现出在药物分子合成方面的应用价值(图3)。

图3 癫痫药物Seletracetam的不对称合成
该研究得到了国家自然科学基金委、中国博士后科学基金项目、湖北省博士后创新岗位项目以及中央高校基本科研业务费专项资金等的资助。